Heatmap and contour plots
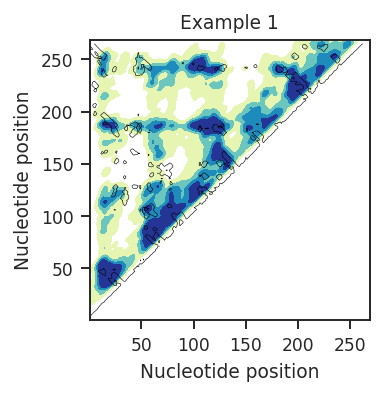
Heatmaps and contour plots are a good way to view inter-nucleotide data when the data are very dense, or when you wish to view inter-nucleotide data density. The x and y axes are nucleotide positions. XY-positions are colored by inter-nucleotide data values or density (heatmap) and overlayed with a contour map. This contour map can show either 3D distance or secondary structure graph path distance (contact distance). These countours will outline areas where nucleotide X is close, in 3D space or secondary structure, to nucleotide Y.
[1]:
import rnavigate as rnav
from rnavigate.examples import rnasep_1
plot = rnav.plot_heatmap(
samples=[rnasep_1],
sequence="pdb",
structure="pdb",
interactions="shapejump",
plot_type="kde",
)
[2]:
help(rnav.plot_heatmap)
Help on function plot_heatmap in module rnavigate.plotting_functions:
plot_heatmap(samples, sequence, structure=None, interactions=None, regions=None, labels=None, levels=None, interpolation='nearest', atom="O2'", plot_type='heatmap', weights=None, rows=None, cols=None, plot_kwargs=None)
Generates a multipanel plot displaying a heatmap of inter-nucleotide
data (nucleotide resolution of 2D KDE) and/or contour map of pdb
distances. Each plot may display a unique sample and/or filtering scheme.
Parameters
----------
samples : list of rnavigate Samples
samples used to retrieve data
sequence : data keyword string, data object, or sequence string
All data are mapped to this sequence before plotting
structure : data keyword string or data object, defaults to None
secondary structure or 3D structure used to plot contour lines
contour lines are drawn according to levels argument
interactions : one of the formats below, defaults to None
format 1 (data or data keyword)
Interactions to plot as a heatmap, no filtering performed
format 2 (dictionary)
e.g. {"interactions": format 1}
additional filtering options can be added to the dictionary
format 3 (list of format 2 dictionaries)
This format allows multiple filtering schemes to be applied,
each will be plotted on a seperate axis
regions : list of lists of 4 integers, defaults to None (no boxes)
each inner list defines two regions of the RNA that are interacting
a box will be drawn around this interaction on the heatmap
e.g. [[10, 20, 50, 60], [35, 45, 70, 80]] draws 2 boxes
the first connects nucleotides 10-20 and 50-60,
the second connects nucleotides 35-45 and 70-80
labels : list of strings, defaults to sample.sample for each sample
Labels to be used as titles, must be same length as samples list
levels : list of floats, defaults to [5] contact distance or [20] 3D distance
contours are drawn separating nucleotides above and below these distances.
If structure is a secondary structure, distance refers to contact distance.
If structure is a 3D structure, distance refers to spatial distance in angstroms.
interpolation : string, defaults to "nearest"
one of matplotlib's interpolations for heatmap (used with imshow)
"nearest" works well for shorter RNAs (under 300 nt)
"none" works well for longer RNAs (over 1200 nt)
atom : string or dictionary, defaults to "O2'"
from which atoms to calculate distances
for DMS reactive atoms (N1 for A and G, N3 for U and C) use "DMS"
use a dictionary to specify a different atom for each nucleotide
e.g. "DMS" == {"A": "N1", "G": "N1", "U": "N3", "C": "N3"}
plot_type : "heatmap" or "kde", defaults to "heatmap"
how to plot interactions data
"heatmap" will plot raw data, each interaction is a pixel in a grid
"kde" will calculate a kernel density estimate and plot 5 levels
weights : string, defaults to None (no weights)
weights to be used in kernel density estimation
must be a column of interactions data
rows : integer, defaults to None (determined automatically)
number of rows of plots
cols : integer, defaults to None (determined automatically)
number of columns of plots
plot_kwargs : dictionary, defaults to {}
Keyword-arguments passed to matplotlib.pyplot.subplots
Returns
-------
rnavigate.plots.Heatmap
object containing matplotlib figure and axes with additional plotting and
file saving methods